Date of Publishing: Sep 01, 2017
DOI: 10.7860/JCDR/2017/29078.10664
Journal of Clinical and Diagnostic Research. 2017 Sep, Vol-11(9): PD17-PD18.
Autoimmune Pancreatitis Type II: First Report from India
Gunjan Desai1, Prasad Pande2, Chandralekha Tampi 3, Prasad Wagle4
Abstract:
Autoimmune Pancreatitis (AIP) presents in two forms - Type I or lymphoplasmacytic sclerosing pancreatitis and Type II or idiopathic ducto-centric pancreatitis (IDCP). AIP II is rare in south Asia and, especially so, in India. Most patients have either Idiopathic Chronic Pancreatitis (ICP) or alcohol related chronic pancreatitis. AIP Type I has been described in India. We herein report a patient who had features of ICP on imaging, for whom surgery was performed to relieve chronic pancreatic pain. However, the pathologic features revealed AIP Type II or IDCP.
CASE REPORT:
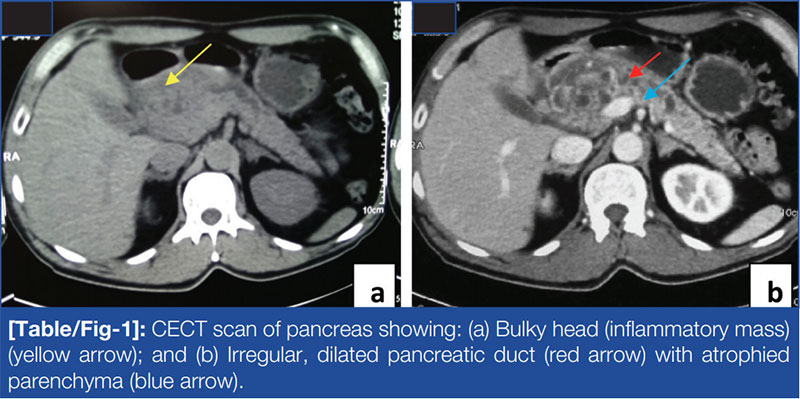
A 53-year-old gentleman had recurrent episodes of dull aching upper abdominal pain over last 5 years increasing on fatty food intake and significant weight loss. There was no history of alcohol abuse. He had marginally elevated alkaline phosphatase. Serum Immunoglobulin G4 (IgG4) was not requested. Contrast enhanced Computerized Tomography (CT) of abdomen showed a 7.5 x 6 cm pancreatic head mass with small cystic areas and 7 mm pancreatic duct [Table/Fig-1] with possible lower common bile duct narrowing. He was diagnosed with ICP and was put on analgesic and pancreatic enzyme therapy.
In view of unremitting chronic pain and imaging findings, patient was offered a plan of either pancreaticoduodenectomy or Frey’s procedure. At surgery, a firm to hard 8 x 6 cm pancreatic head mass was seen from which multiple frozen biopsies were taken, all of which were benign. A Frey’s procedure was performed. The bile duct was exposed in pancreatic bed. Cholecystectomy was done and a probe passed via cystic duct into the common bile duct entered duodenum without resistance obviating the need for biliary enteric anastomosis.

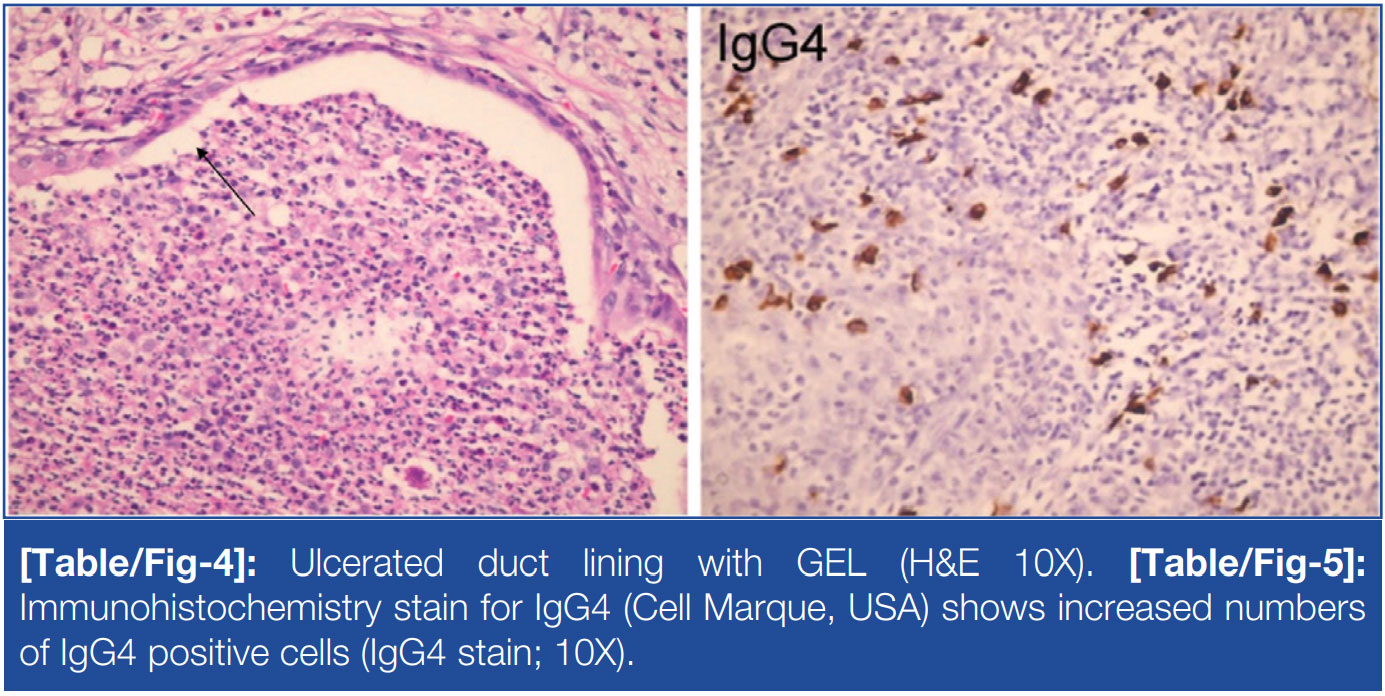
Histopathology showed features of chronic pancreatitis, a dilated pancreatic duct and ductocentric inflammation. The mucosal lining showed hyperplasia, ulceration, foci of squamous metaplasia and infiltration by lymphocytes, plasma cells and neutrophils [Table/ Fig-2]. The lymphoplasmacytic inflammatory infiltrate was seen predominantly around the large and medium sized ducts [Table/ Fig-3]. Intraductal neutrophilic abscesses were seen [Table/ Fig-4]. No phlebitis, storiform fibrosis, fat necrosis, calcification, malignancy or collagenisation was seen. Immunohistochemistry
stains for IgG4 showed slightly increased numbers of IgG4 positive cells [Table/Fig-5]. Diagnosis of AIP II was made. The patient was asymptomatic.
DISCUSSION:
AIP is a rare aetiology of chronic pancreatitis [1]. Type II AIP is also called idiopathic ducto-centric pancreatitis [2] and can be confused with malignancy [3]. The International Consensus of Diagnostic Criteria (ICDC) is used for diagnosis [4]. Endoscopic Ultrasound (EUS) guided biopsy is helpful in doubtful cases [1]. Granulocytic Epithelial Lesion (GEL) is the diagnostic characteristic of AIP Type II [5]. In case of doubt, surgery can exclude malignancy and treat symptoms [3]. We report the above case of AIP Type II which to our knowledge is the first case from India
AIP is a rare form of chronic pancreatitis with a different genetic and demographic predisposition, aetiopathogenesis and extrapancreatic manifestations [1]. It was first described in 1961 by Henry Sarles [6]. Yoshida in 1995 coined the term AIP [7]. Of the two types, Kamisawa suggested that Type I AIP is a part of a multisystem IgG4 related disease [8]. Type II AIP, on the other hand, is a pancreas specific disease with no gender or age predilection, normal serum IgG4 levels and histology of ducto-centric pancreatitis [2]. It is more common in Europe and America (USA) [4,5]. It often presents with abdominal pain, obstructive jaundice, new onset diabetes, weight loss, abdominal mass and exocrine pancreatic insufficiency [2,4,6]. The only extrapancreatic manifestation is ulcerative colitis [1]. Our patient had chronic abdominal pain and weight loss.
Diagnostic criteria for AIP Type II have evolved from the Japanese group based on histology, imaging, associated autoimmune disorders and pancreatic function status to Korean group based on imaging, histology, serology and response to steroids to the well known HISORt (Histology, Imaging, Serology, other Organ Involvement and Response to therapy) criteria of Mayo clinic and finally, ICDC which includes imaging, histology, serology and other organ involvement to define the disease [4,6].
Imaging characteristics of AIP are diverse [3,5,7]. On CT-scan, it can diffusely involve the pancreas leading to a sausage shaped pancreas with a hypoattenuating rim or can be focal or multifocal involvement of pancreas wherein the involved area is iso to hypoattenuating in pancreatic phase and mildly hyper enhancing in hepatic phase [3]. This enhancement in hepatic phase is not seen in malignancy. Also, it is hypo to isointense on T1 and T2 images with delayed enhancement on magnetic resonance imaging scan. Apparent Diffusion Coefficient (ADC) values are significantly lower in AIP. The ADC values in AIP are typically below 1.075*10 whereas they are higher in cancer.
Pancreatic duct involvement shows more than one third of the duct being involved in single or multifocal strictures [3,5]. There is no ductal dilatation upstream of the strictures and this helps in differentiating AIP from carcinoma pancreas. Our case is an exception to this rule and shows ductal dilatation upstream to the stricture. Also, atrophy of the parenchyma upstream of the involvement is uncommon in AIP whereas it is present in carcinoma [1,7].
Histologically, AIP II does not have IgG4 positive cells, has no obliterative phlebitis and storiform fibrosis [2]. It has neutrophilic infiltrates within the duct lumen. GEL is pathognomonic of AIP II, which causes immune complex mediated pancreatic duct destruction and Complement Deposition (C3c) in pancreatic duct and acinar basement membrane [5]. The histology showed these features in our patient.
Serologically, IgG4 levels are < 2 times normal [5]. Ca 19.9 can be elevated. EUS guided biopsy would be helpful in doubtful cases by excluding malignancy [1]. If the diagnosis is established without surgery, 30-40 mg/day prednisolone for four weeks followed by taper at 2.5-5 mg/week till 5 mg/day maintenance dose for six months achieves remission in 92% patients [1]. Some studies in Asia have advised maintenance doses upto three years whereas, studies in USA and Europe have not advised maintenance therapy [3,4]. We managed the patient with surgery and without any steroid therapy.
Relapse in AIP II is rare as factors increasing relapse rates such as increased IgG4 levels and extrapancreatic manifestations are not present in AIP II [1]. Clinical and/or radiological relapse mandate further treatment. Serological relapse does not require therapy. Drugs in refractory AIP include steroids, azathioprine, methotrexate, rituximab amongst others. The long term prognosis is not yet defined [1,7].
Our case is the first reported case of AIP II from India with a history of chronic pancreatitis with head mass and unremitting pain, hence, managed with a Frey’s procedure.
CONCLUSION:
AIP II is rare in India, often confused with malignancy and difficult to diagnose without histology. Further studies are required to establish treatment protocols for patients with AIP II managed with or without surgery
REFERENCES:
- Salem A, Hamouda D, Parian A. Review article: diagnosis and management of Igg4 autoimmune pancreatitis. J Pancreas (Online). 2015;16(4):326-34.
- Pezzilli R. Pathophysiology of autoimmune pancreatitis. World Journal of Gastrointestinal Pathophysiology. 2014;5(1):11.
- Takahashi N. CT and MR Features of Autoimmune Pancreatitis. The Pancreapedia: Exocrine Pancreas Knowledge Base. 2013. doi:10.3998/panc.2013.15.
- Shimosegawa T, Chari S, Frulloni L, Kamisawa T, Kawa S, Mino-Kenudson M, et al. International consensus diagnostic criteria for autoimmune pancreatitis. Pancreas. 2011;40(3):352-58.
- O’Reilly DA, Malde DJ, Duncan T, Rao M, Filobbos R. Review of the diagnosis, classification and management of autoimmune pancreatitis. World J Gastrointest Pathophysiol. 2014;5(2):71-81.
- Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas-an autoimmune pancreatic disease? Am J Dig Dis. 1961;6:688-98.
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40(7):1561-68.
- Kamisawa T, Chari ST, Lerch MM, Kim MH, Gress TM, Shimosegawa T. Recent advances in autoimmune pancreatitis: type 1 and type 2. Gut. 2013;62(9):1373-80.
PARTICULARS OF CONTRIBUTORS:
1. Registrar, Department of Gastrointestinal Surgery, Lilavati Hospital and Research Centre, Mumbai, Maharashtra, India.
2. Registrar, Department of Gastrointestinal Surgery, Lilavati Hospital and Research Centre, Mumbai, Maharashtra, India.
3. Consultant, Department of Pathology, Lilavati Hospital and Research Centre, Mumbai, Maharashtra, India.
4. Consultant, Department of Gastrointestinal Surgery, Lilavati Hospital and Research Centre, Mumbai, Maharashtra, India.